


作品描述:
Best practice of MIQE 投稿
1、结合实例,分析MIQE 指南的具体应用
2、分享如何在MIQE 指南的指引下开展qPCR 实验
(包括但不限于,实验设计中取样大小的估计、设置NTC、NRT 、阳性对照以及复孔、RNA 质量评估、Primer 设计与扩增子、qPCR assay 的优化与验证、内参基因的筛选和验证、合理的数分析流程等等),具体模板如下:
Sample Type and source:
Materials and Methods: including Target gene description, Primer design, reverse
transcription, qPCR reagent and consumable, Assay optimization and validation, reference gene
selection and validation, data analysis
Brief introduction:
The experiment was based on genome synthesis of a specific genomic region of an important industrial workhorse gram-positive strain Corynebacterium glutamicum. Briefly, the genomic sequence of C. glutamicum ATCC 13032 was redesigned, including (1) decoupling overlapping genes, replacing TAG stop codons to TAA, (2) inserting 34-bp loxPsym sites into 3 bp downstream of the stop codons of monocistronic genes or the last genes in operons, and (3) synonymous substitutions of PCRtags for distinguishment of synthetic sequences from the wild-type ones. While these changes in DNA sequences should not alter sequences of relevant proteins, they might have mild or significant influences on expression of genes.
To better investigate the effects of these DNA alterations (especially decoupling overlapping genes) on transcription, qRT-PCR was performed in our previous work. The following paragraphs were to present the experience of conducting qPCR experiments following the guidelines of MIQE (Minimum Information for Publication of Quantitative Real-Time PCR Experiments) guidelines, and publishing experimental data and academic papers compliant with the MIQE guidelines.
Experimental design:
Control group: the wild-type C. glutamicum ATCC 13032
Experimental group: the strain with synthetic sequence replacement, defined as semi-synCG-A1;
Samples:
The inoculation of strains, total RNA extractions and qRT-PCR experiments were performed in three biological replicates, respectively.
Both strains C. glutamicum ATCC 13032 and semi-synCG-A1 were grown overnight in BHIS medium in 30 °C to exponential phase (OD600 = 0.6~0.8). Then 1 mL of the cells was collected by centrifugation.
Nucleic acid extraction:
Total RNA extraction was performed as described. The pellets described above were used for RNA extraction using RNeasy Mini Kit (QIAGEN, Germany). Beads and vortex were used for lysis of cells. The RNase-free tubes and pipets (Axygen) were just unpacked before experiments. The quality and concentration of RNA was determined on Nanodrop (ThermoFisher). The samples with OD260/OD280 > 2.0 were chosen.
Reverse transcription:
After extraction, Reverse transcription was preformed using a PrimeScript™ RT reagent kit with a gDNA Eraser (TaKaRa, China). Following manufacturer’s instruction, the RNA was diluted to no more than 1 ng in 20 μL of reverse transcription reaction. In each step, gentle pipetting was used instead of vortex for better efficiency. The yield of cDNA was 1300-1400 ng/μL.
qPCR target information:
Detailed information of names of target genes, primers used for qPCR have been posted on the published article. Briefly, amplicons were mostly around 80-150 bp, but can be longest as 250 bp. For consistency of amplification efficiency, the qPCR primers were designed on specific genomic region that was subjected to redesign, while the primers were to probe the genomic sequence that was not altered on either version, so that the efficiency was the same on either group of strains. The length of primers was 17-22 nt, with 45-55% GC, Tm 54-58°C on Snapgene. Specificity screen was performed on both BLAST, Oligo and Primer Premier 5 software to avoid situations of hairpin, dimer or false priming.
For internal controls, 16s rRNA and NCgl2772 genes were chosen.
Oligonucleotides were purchased from Sangon Biotech (Shanghai, China).
qPCR oligonucleotides:
List of primers used was provided on the supplementary file that can be assessed.
qPCR protocols:
qPCR was performed using SYBR® Premix Ex Taq™ II (TaKaRa, China) on CFX96 Real-Time PCR Detection System (Bio-Rad). The template cDNA was diluted to ~40 ng/uL with ddH2O. The total volume of each reaction was 25 μL, and the final concentration of primers and template was 0.2 μM and no more than 80 ng. Thermocycler conditions were set as follows: 95°C for 30s, 40 cycles of (95°C for 5s, 60°C for 30s), and a final procedure for melt curve (95°C for 5s, 65 to 95°C for 5s).
qPCR optimization and validation:
Same volume of ddH2O was used as NTC.
To minimize errors on operation, when preparing qPCR reaction mix, the templates were always mixed with polymerases. The primers were always mixed prior to final mixing of all the components. Automatic pipettor (Eppendorf) was used for reaction mixture.
Data analysis: Original Cq value was given by the Bio-Rad CFX Mastero software and manually checked. Gene expression was quantified using the comparative threshold (ΔΔCt) method.

Results:
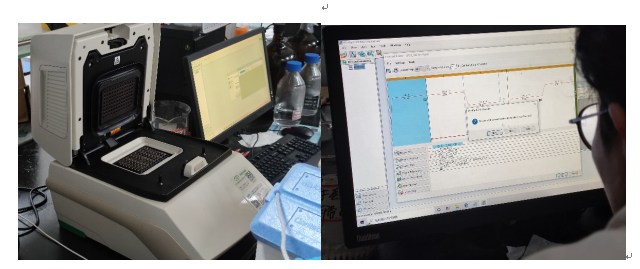
Figure 1 Operation on Bio-Rad CFX96 and the Bio-Rad CFX Mastero software
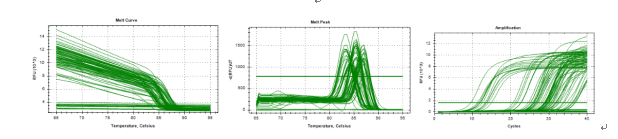
Figure 2 Results of melt curve, melt peak and amplification cycles during several qPCR reactions generated by Bio-Rad CFX Mastero software
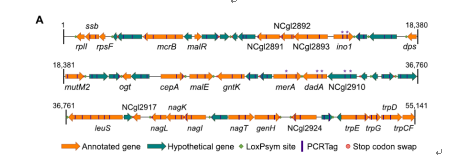
Figure 3 Detailed genomic replacement on the defined region (published)
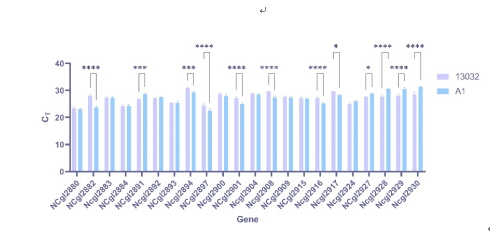
Figure 4 CT value of the chosen genes comparing to 16S rRNA gene
Short results summary
Generally, the amplification showed moderate efficiency. A significant proportion (13/22) of the redesigned genes were differentially expressed (with log2 fold changes >1 or <−1), and 11/14 of the decoupled genes (some with synonymous recoding and/or loxPsym insertions) were differentially expressed compared to those of the wild type. In contrast, only 2/8 of the genes without decoupling (some with synonymous recoding and/or loxPsym insertion) showed differential expression. These results indicated that gene decoupling has more profound impacts on transcription than synonymous recoding and loxPsym insertions. However, the expression difference between decoupled and non-decoupled genes was not statistically significant (P = 0.242, t-test).
It seems that decoupling of the overlapping genes had more impacts on the transcription than on the phenotype in the current 55.1 kb version of the synthetic C. glutamicum strain. In this work, ∼27% of the genes (14/52) were decoupled. These results have been reported on our previous publish article.
For further investigation of these genes, detailed investigation of transcription start and end could provide insight in the cause that have led to diverse gene expression patterns.
For preparation of the synthetic version of the genome of semi-synCG-A1, massive efforts have been made including molecular cloning for assembly of fragments, electro-transformation and recombineering on C. glutamicum strains and phenotyping and genotyping analysis, which we have reported. Bio-Rad apparatus have facilitated PCR amplification of fragments, Gibson Assembly for construction of the small plasmid, electroporation, PCR gel electrophoresis and imaging.
In all, the Bio-Rad equipment has provided significant help in convenient and effective screening and analyzing of investigation of transcriptional expression of genes of interests, fulfilling genome synthesis in C. glutamicum, a promising industrial prokaryotic strain.
Additional points 加分项
Journal SCI Publication (optional): Genomic Iterative Replacements of Large Synthetic DNA Fragments in Corynebacterium glutamicum
Journal link (optional): https://pubs.acs.org/doi/10.1021/acssynbio.1c00644
(optional)qPCR experiment tips:
− 符合 MIQE 指南的实验技巧,如设计引物以跨越外显子-外显子交界处、如何避免为具有二级结构的区域设计引物、利用温度梯度优化protocol 等。
For qPCR of bacterial samples, several principles of qPCR primer design were effective. Specifically, (1) Amplicons were mostly around 80-150 bp. (2) The length of primers was 17-22 nt, with 45-55% GC, Tm 54-58 °C and ΔTm≤2 °C on Snapgene. (3) Forward primers were better in the 100-300bp from the 5’ of the ORF. (4) Avoid situations of hairpin, dimer or false priming if possible.
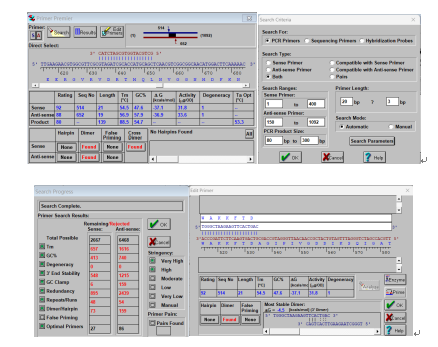
When using Primer Premier 5 for design of qPCR primers, “Search” button can be used to automatically search for appropriate primers with preferred criteria. The follow was shown as an example.
(optional)Other additional information
− 如涉及qPCR 实验,需提供必要的qPCR 实验细节
Relevant experiment settings and results were provided above.
− 使用Bio-Rad 的CFX qPCR 仪、数据分析软件CFX Maestro、试剂、塑料耗材,并提供相应的证据,如图片
Relevant figures were provided in Figure 1.
问题调研:
问题1:
你在了解MIQE 指南以及相关的学习资料之后,对照自己的qPCR 实验流程,哪些实验步骤之前忽视了,或者哪些实验环节做的不够充分?
回答1: 对实验中的这些细节:amplicon length,specificity (gel), LOD assay.
忽略了这些信息:作为对试验样品质量的检测,以及直观展示扩增设计,应该对应展示扩增子长度和RNA跑胶结果。
不够充分的环节:由于实验只需要对基因表达量相对定量,对绝对定量所需的标准曲线(标准方程、扩增效率和相关系数)的分析并不算完备(但这些信息从Bio-Rad CFX 下机后可以自动生成和查阅)。
问题2:你在qPCR 实验中遇到的问题有哪些?如果使用MIQE,是否可以帮助你解决这些问题?
回答2:MIQE可以解决高效一些基本问题,如引物设计、实验方案和可重复性、数据处理原则。但如果涉及具体的菌株具体研究的内容,还是需要特定的试验方案才能解决问题。如本实验所提及的原核细菌的qPCR,有许多特化的实验方案,如RNA提取时细菌裂菌方法和提取试剂的选择(及对最终结果的影响),qPCR引物设计时参考的基因组序列(上文提及),甚至是不同机型对不同反应体系的影响,这些暂时无法仅通过查阅MIQE来解决。
问题3:
你们实验室是否需要qRT-PCR 相关的培训,如有,希望在2024 年什么时间段?
回答3: 需要;可行的时间是2024学年下学期开学及2024年国庆后。
以上加分项适用于所有投稿作品





